I. Molecular evolution
Why life on Earth is the way it is? Molecular evolutionary study can help elucidate many aspects of this question. Study of molecular systems that constitute a living cell unravels lots of extraordinary, unexpected "approaches" nature takes in order for life to adapt and proliferate.
|
Amino acid binding sensor found across the Tree of Life
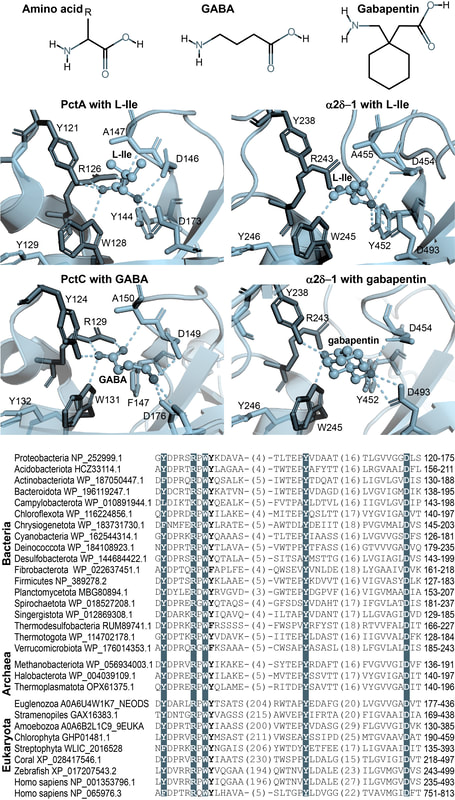
Amino acids are the building blocks of life. However, despite their common structure and indispensable nature no common mechanism of amino acid binding is known. Does a universal mechanism of amino acid binding exist? My research showed that it indeed does.
While studying bacterial chemoreceptors I have discovered a universal amino acid binding motif present in all cellular life forms, from bacteria to humans (link to the paper). This motif is present in a subclass of a sensor domain called dCache_1. In Bacteria and Archaea this motif exclusively binds amino acids, including gamma-aminobutyric acid (GABA), and it is present in all major signal transduction proteins. In humans, I found this motif in α2δ subunit of high voltage-gated calcium channels that are implicated in numerous diseases including neuropathic pain and neurodevelopmental disorders, and in a recently characterized CACHD1 protein that has been suggested to play a modulatory role in low voltage-gated calcium channels. Identification of the amino acid motif allowed me to get deep insights into the structure of α2δ and CACDH1 proteins and uncover extraordinary evolutionary events that gave rise to these eukaryotic proteins. It is truly amazing to "witness" how bacterial proteins shaped a particular aspect of the nervous system in Metazoa. Site-directed mutagenesis performed with bacterial and mammalian proteins showed that mutation of key positions of the motif leads to drastic reduction in binding of amino acids and GABA-derived drugs. My study with colleagues strongly suggests that GABA-derived drugs bind to the same motif in human α2δ subunits that binds natural GABA ligands and alpha amino acids in bacterial signal transduction proteins. The exact location of the binding site on a target protein and the mechanism of binding enable future improvements of drugs targeting pain and neurobiological disorders. Discovery Gumerov V.M., Andrianova E.P., Matilla M.A., Page K.M. , Monteagudo-Cascales E., Dolphin A.C., Krell T., Zhulin I.B. (2022) Amino acid sensor conserved from bacteria to humans. PNAS, 119 (10) e2110415119. |
|
Evolution of bacterial chemoreceptors
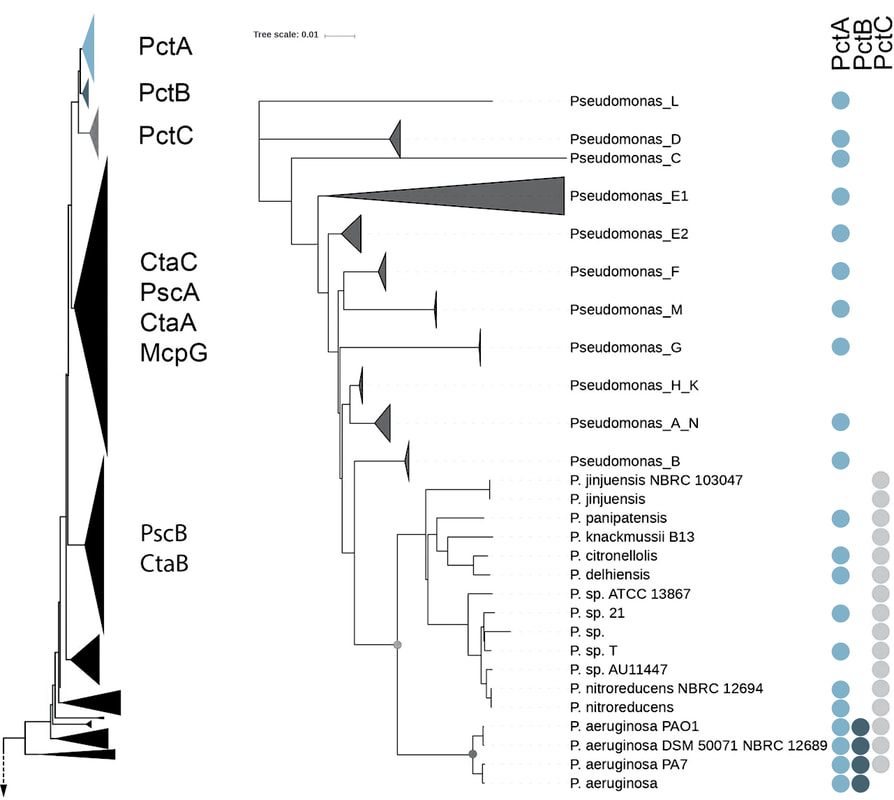
Bacteria need to constantly adapt to changing environmental conditions to ensure survival. Among the different adaptational strategies is the ability of many bacteria to perform chemotaxis, a behavior based on the action of chemosensory pathways. To recognize the variety of chemicals, many bacteria have evolved a large number of different chemoreceptors. Ligand specificity for the vast majority of them remains unknown. Even less is known about how particular sets of chemoreceptors within a given species have evolved.
To develop a framework for addressing these questions I have studied a genome of the pathogenic bacterium, Pseudomonas aeruginosa PAO1. Its genome encodes 26 chemoreceptors. One of these chemoreceptrs, PctA, has a broad ligand range and responds to most of the proteinogenic amino acids, whereas two other paralogs, PctB and PctC, have a much narrower range and show strong ligand preference for L-glutamine and gamm-aminobutyrate, respectively. Using comparative genomics approaches, I showed that these receptors are paralogs (link to the paper): pctA gene duplication in the common ancestor of the genus Pseudomonas led to pctC, whereas pctB originated through another, independent pctA duplication in the common ancestor of P. aeruginosa. These results allowed me to establish that the broad-range amino acid chemoreceptor was evolutionary older, and chemoreceptors that complemented “missing” amino acid sensing abilities arose later in specific Pseudomonas lineages. Furthermore, I have determined that this group of receptors rapidly evolves and obtained the first glimpse of the universal amino acid binding motif. Gavira J.A., Gumerov V.M., Rico-Jimenez M., Petukh M., Ortega A., Matilla M.A., Zhulin I.B., Krell T. (2020) How Bacterial Chemoreceptors Evolve Novel Ligand Specificities. mBio, 11: e03066-19 |
|
A new class of metallo-β-lactamases
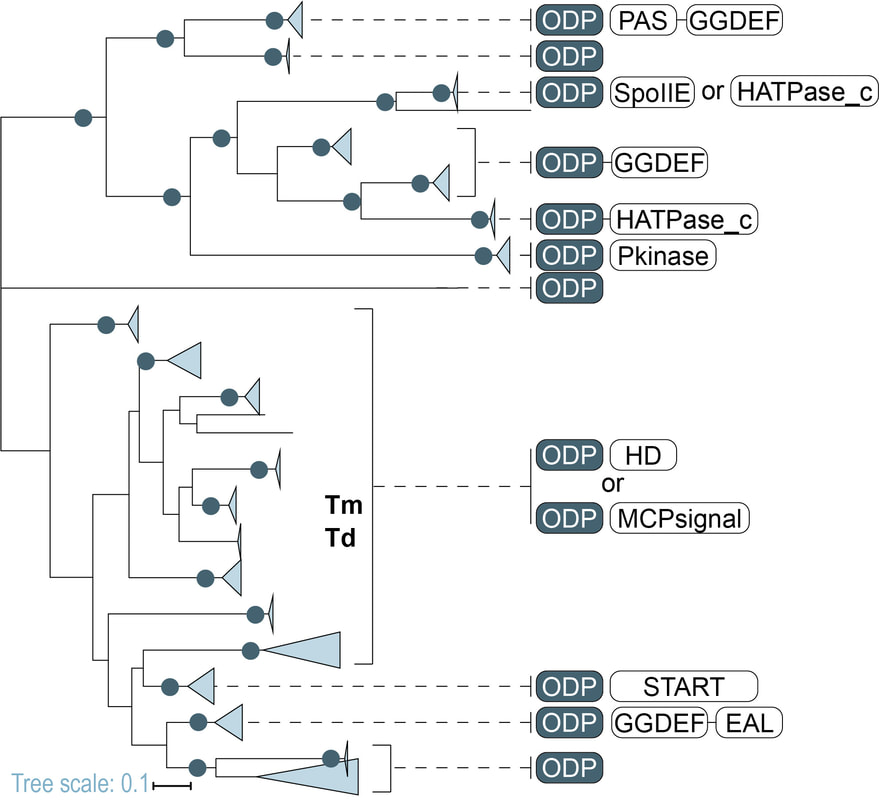
Many bacteria contain cytoplasmic chemoreceptors that lack sensor domains. In the collaborative work we demonstrated that a subclass of such cytoplasmic receptors found in 8 different bacterial and archaeal phyla genetically couple to a specific group of metalloproteins.
While studying these metalloproteins I discovered that they all have the same unique metal-binding motif and represent a new previously unknown class of metallo-β-lactamases (link to the paper). They are related to the family of flavodiiron proteins (FDPs) and serve as oxygen and/or nitric oxide reductases. I have established that this protein co-evolves with specific cytoplasmic chemoreceptors and was recruited to signal transduction as sensor module. Based on the experiments with the human pathogen Treponema denticola the protein was found to bind both iron and oxygen and oxygen binding is reversible. Furthermore, the protein was found to mediate chemotaxis toward iron and away from oxygen. Muok A.R., Deng Y., Gumerov V.M., Chong J.E., DeRosa J.R., Kurniyati K., Coleman R.E., Lancaster K.M., Li C., Zhulin I.B, Crane B.R. (2019) A di-iron protein recruited as an Fe[II] and oxygen sensor for bacterial chemotaxis functions by stabilizing an iron-peroxy species. PNAS, 116: 14955-14960. |

|
Molecular evolution of the NusG transcription factor paralog RfaH
Housekeeping regulators and their specialized paralogs are the only universally conserved transcription factors. They are represented by well-studied NusG and RfaH. Despite their ubiquity, little information was available on the evolutionary origins, functions, and gene targets of the NusG family members. In contrast to NusG which is universally present in prokaryotes RfaH is mostly limited to Proteobacteria and lacks common gene neighbors. It activates only a few xenogeneic operons that are otherwise silenced by NusG and Rho.
Phylogenetic reconstructions revealed extensive duplications and horizontal transfer of rfaH genes. My computational work helped establish that NusG is not only ubiquitous in Bacteria but also common in plants and Chromista, in which it likely modulates the transcription of plastid genes (paper). Markov clustering and phylogenetic analysis of RfaH sequences allowed to propose a step-wise conversion of a NusG duplicate copy into a sequence-specific regulator which excludes NusG from its targets but does not compromise the regulation of house-keeping genes. Gene duplication and lateral transfer gave rise to a surprising diversity within this family of transcription factors. Wang B., Gumerov V.M., Andrianova E.P., Zhulin I.B., Artsimovitch I. (2020) Origins and Molecular Evolution of the NusG Paralog RfaH. mBio, 11: e02717-20. |
II. Computational pipelines and databases
Computational tools and databases are the hallmarks of bioinformatics and computational biology. They allow for large-scale studies that lead to the discovery of new phenomena and initiate new research directions.

|
Microbial Signal Transduction database (MiST)
With the team of talented bioinformaticians I have created a comprehensive online database of microbial signal transduction systems called MiST (https://mistdb.com/). The database includes more than 125,000 sequenced genomes and their signal transduction profiles and 516 million genes. The database has been used in numerous projects around the world.
Gumerov V.M., Ortega D.R., Adebali O., Ulrich L.E., Zhulin I.B. (2020) MiST 3.0: an updated microbial signal transduction database with an emphasis on chemosensory systems. Nucleic Acids Res., 48: D459-D464. |
|
Computational platform for protein evolution and function analysis (TREND)
Computational analysis of protein evolution and function can lead to deep insights and unexpected discoveries. However a comprehensive analysis is extremely time-consuming and error-prone. To help biologists perform evolutionary analysis more efficiently I have created a computational platform called TREND (http://trend.evobionet.com/). The platform automates many steps in the computational analysis of proteins and puts all the information obtained into a phylogenetic context.
Gumerov V.M. and Zhulin I.B. (2020) TREND: a platform for exploring protein function in prokaryotes based on phylogenetic, domain architecture and gene neighborhood analyses. Nucleic Acids Res., 48: W72-W76. |
III. Extremophilic Bacteria and Archaea
It has been estimated that bacteria account for ~60% of Earth’s biomass. Many bacteria cause disease in humans and other animals. The analysis of whole genomes, boosted by next-generation sequencing, has had profound effects on our understanding of bacteria and archaea.
|
Genomes
Comparative genomics and phylogenetics help discover numerous new phenomena: regulatory elements, protein domains, recruitment of domains to new biological processes, viral elements, sequence motifs, etc. Microorganisms that live in extreme environments have developed various unique molecular strategies to adapt to such conditions. These mechanisms and in particular enzymes encoded in the genomes of these organisms find important industrial, medical and research application (for example, thermostable Taq polymerase)
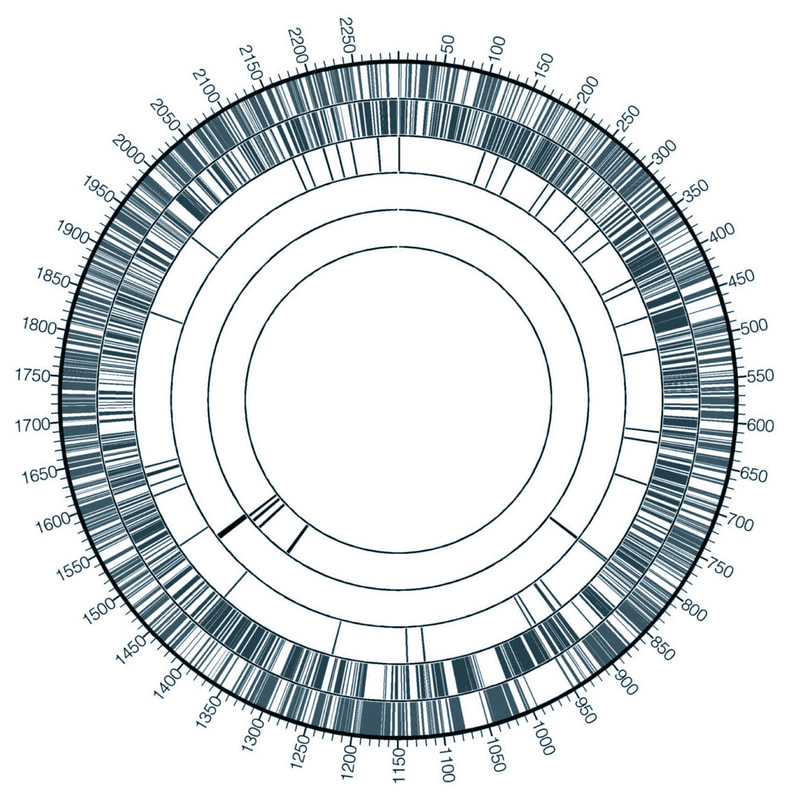
As a graduate student I went on an expedition to Kamchatka to collect samples from the habitats with extreme conditions to study how genomes of the extremophilic microorganisms are organized. I studied genomes and encoded metabolic pathways of archaea and bacteria isolated from such harsh environments as boiling acidic springs (ref1, ref2, ref3, ref4) and hypersaline soda lakes (ref5, ref6) and uncovered unusual metabolic adaptations of these organisms. In one of such projects my analysis enabled identification and description of two new classes of bacteria, Chitinivibrionia (ref5) and Chitinispirillia (ref6), within the Fibrobacteres phylum. Genomes deposited to NCBI: Vulcanisaeta moutnovskia 768-28 Chitinivibrio alkaliphilus ACht1 Chitinispirillum alkaliphilum Acht6-1 Pyrobaculum ferrireducens 1860 Thermoproteus uzoniensis 768-20 Fervidicoccus fontis Kam940 |
|
Enzymes
Enzymes from bacteria and archaea have a wide application in various industrial processes. Extremophilic microorganisms represent a rich source of enzymes with unusual properties. Enzymes isolated from extremophyles, or extremozymes, are able to catalyze chemical reactions under extreme conditions, similar to those found in industrial processes. Due to their stability under such conditions, extremozymes offer new catalytic alternatives for industrial applications.
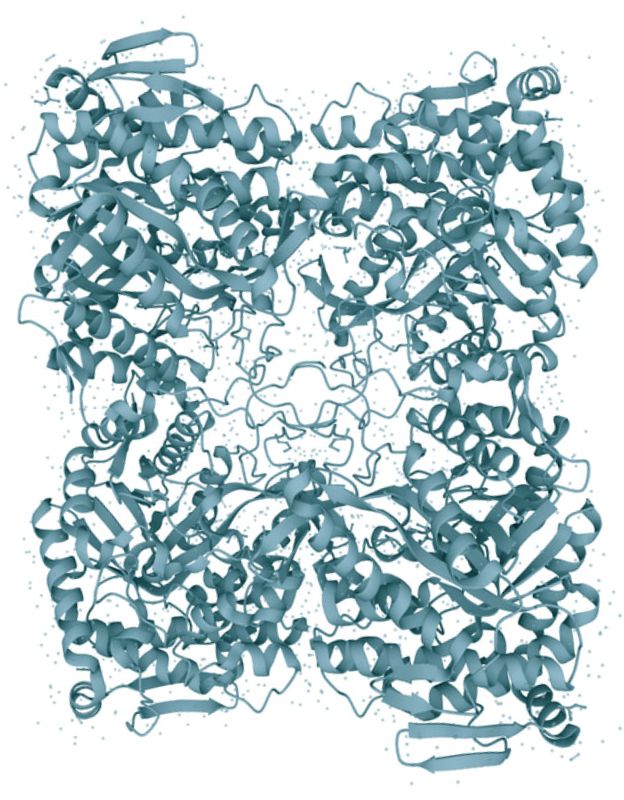
My analysis of extremophilic archaea resulted in identification and biochemical characterization of several thermostable enzymes with unique properties: multifunctional beta-glycosidase (ref1), lipase (ref2), alcohol dehydrogenase (ref3, ref4, ref5), superoxide dismutase (ref6), M42 aminopeptidase (ref7), and prolidase (ref8). Structures deposited to PDB: Beta-glycosidase from Acidilobus saccharovorans: - in complex with Tris: PDB 4HA3 - in complex with glycerol: PDB 4HA4 Alcohol dehydrogenase from Thermococcus sibiricus: PDB 3TN7 Aldehyde dehydrogenase from Pyrobaculum ferrireducens 1860: - in apo form: PDB 5EEB - in complex with NADP+: PDB 5F2C, 4NMJ, 4NMK, 4H73 Iron superoxide dismutase from Acidilobus saccharovorans: 4FFK Prolidase from Thermococcus sibiricus: PDB 4FKC |

|
Microbial communities
Investigation of the microbial communities associated with thermal ecological niches is of great interest for evolutionary biology as many microorganisms living in these conditions belong to ancient evolutionary lineages of thermophilic bacteria and archaea.
Hot springs have been investigated since the XIX century, but isolation and examination of their thermophilic microbial inhabitants did not start until the 1950s. Many thermophilic microorganisms and their viruses have since been discovered. 16S rRNA-based studies subsequently revealed that microbial diversity was much broader than suggested by culture-dependent techniques. I investigated microbial communities of hot and acidic springs of Kamchatka volcanic regions. One of my findings is that phylogenetically diverse microbial communities consisting of various metabolic groups of prokaryotes can thrive even under the most extreme conditions of low pH and high temperatures (ref1, ref2, ref3). Remarkably, in the thermal groundwater of the Uzon volcano crater Archaea are the predominant component of microbial communities (ref4)! Another interesting finding is that in several acidic thermal pools Nanoarchaeota - an obligate symbiont on another archaeon Ignicoccus - comprise a significant part of the communities (ref1). It is interesting to note, that the genome of this organism lacks essentially all the genes required for the synthesis of nucleotides, amino acids, cofactors, and lipids, but encodes all the genes necessary for repair and replication. |